 |
|
Content:
Research in our group covers two areas, molecular encapsulation and de novo proteins. The former area involves a study of templation effects in the assembly/construction of molecular prisons (small molecules imprisoned inside larger molecules). We design, synthesize, and characterize complex organic molecules that have well-defined 3-D structure. We have revealed unprecedented recognition to be involved in this process, including a million-fold range in template effects. Such sensitivity in recognition is of paramount importance to supramolecular chemistry and indeed to much of biological chemistry. We coined the term "template ratios" to characterize this template process. Potential applications of this work include drug delivery devices, sensors, and protectors of dyes. In the de novo protein work, we have created among the most native-like non-natural proteins ever reported: Combination of a non-natural organic scaffold with designed peptides renders a protein with structural properties on par with natural proteins. Such work represents a significant breakthrough in the protein folding problem, as it is now possible albeit in a limited fashion to design a protein from scratch. Applications include creation of new catalysts that are more robust than natural enzymes and can be tuned to non-natural substrates. The significance of templation is manifold. A great number of reviews have been written on the subject, including two recent books on templation in organic synthesis. DNA is replicated using itself as a template. Messenger RNA is transcribed from the DNA template. Translation of RNA into proteins occurs via use of the RNA as a template. Crystal growth is essentially a templation process where nucleation provides a template. New types of molecules can be created using templates. Pedersen created crown ethers via a template synthesis and was awarded the Nobel Prize for his achievement. Reaction yields can be significantly enhanced using templates. Product distributions can be manipulated using templates. Reaction rates can be increased using templates. Unwanted side products can be reduced using (negative) templates. In short, templation involves recognition processes that are ubiquitous in biology, organic synthesis, and materials science. Understanding of such processes are of fundamental interest and widespread utility. Templated Synthesis of Carceplexes, Hemicarceplexes, and Capsules, Makeiff, D.; Sherman, J. C. in Templated Organic Synthesis, Stang, P. J., Diederich, F. N., Ed., VCH, 1999.
Our Early Templation Work (Bob Chapman) 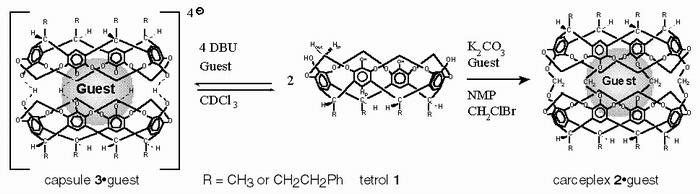 A carceplex is a molecule that permanently incarcerates a guest molecule within its confines by virtue of having small pores relative to the size of the guest. Cram proposed such a species in 1983 and reported the first carceplex, an insoluble solid, in 1985. Subsequently, the first fully characterized soluble carceplex (2oguest) was reported in 1989 (done by John as a graduate student with D. J. Cram): reaction of tetrol 1 with CH2BrCl in the presence of base yields carceplex 2oguest (tetrol 2 is available in 10 gram quantities in a four step synthesis). Carceplex 2oguest will only form in the presence of a suitable guest molecule, which acts as a template. Our group embarked on an investigation of this template effect. Competition of one guest molecule against another revealed the "template ratios" for 34 guest molecules. These ratios were determined by measuring the product ratios (via integration of NMR signals) from competition reactions in which carceplex mixtures are formed in the presence of two guests. In most cases involving templated synthesis, the same product is formed by two templates (e.g., 18-crown-6 from Na+ and K+) and it is difficult to determine how much product is formed by each template. The beauty of our system is that although essentially the same product is formed by each template, the products are "tagged" by the templates, as one template molecule is entrapped in each carceplex. Thus the template ratios can be determined easily and accurately, and large effects can be revealed (in contrast, merely measuring yields in the presence of different templates gives only indirect, qualitative information). Template ratios reflect the relative rates of the guest-determining step (GDS, the step beyond which no guest exchange occurs) of the reaction. The template ratios range one million-fold (i.e., the GDS is 106 times faster in the presence of the best versus the poorest template), which is remarkable when one considers that the guests are all neutral and that no metal coordination is involved; thus, weak noncovalent interactions are responsible for a tremendous range in the rates of the GDS. Moreover, yields were as high as 87%, and a 75% yield was obtained in the presence of only a stoichiometric amount of guest. Thus there is an unusually marked template effect at play, where seven molecules come together and eight covalent bonds are made with high efficiency. Chapman, R. G.; Chopra, N.; Cochien, E. D.; Sherman, J. C. J. Am. Chem. Soc. 1994, 116, 369-370.
More on Our Early Template Work (Bob Chapman, Janet Fraser) Direct investigation of complexation of guests by tetrol 1 showed that two bowls wrap around guests in the presence of base to form reversible capsule 3oguest, according to 1H NMR spectra in CDCl3, electrospray mass spectra, and a crystal structure. The relative thermodynamic stabilities of the capsules (in the presence of different guests) correlate with the kinetic template ratios obtained in the formation of carceplex 2oguest; this trend has also been reproduced computationally. Thus, capsule 3oguest is a good model for the transition state of the GDS in the formation of carceplex 2oguest.  Thermodynamic analysis of the above capsules suggests that they are highly sensitive to small perturbations in the guests, which agrees well with other data. For example, methyl acetate is superior to ethyl acetate as a template in forming carceplex 2oguest by a factor of 10,000. And 1,4-dioxane is 1400 times better than 1,3-dioxane. Rotation of guest about the C2 axes of the host vary from slow on the NMR timescale at 175 °C to fast on the NMR timescale at -70 °C, depending on the guest. Pyrazine manifests a 19 kcal/mol energy barrier to such rotation, which implies a very tight fit and strong recognition of pyrazine's shape and electronics: this is the barrier for exchanging the positioning of pyrazine's nitrogens from the host's "equator," A, to it "poles," B. This is arguably a very subtle change, but the cage is highly sensitive to it. We have also found that the bowls are twisted with respect to one another and that the interconversion of these "twistomers" can vary by 3.9 kcal/mol, depending on the guest. Lastly, 1,4-dioxane and 1,4-thioxane suffer 1.6 and 1.8 kcal/mol (respectively) constraints to ring-flipping when incarcerated as 2oguest. For 1,4-dioxane, the same 1.6 kcal/mol effect was observed when encapsulated in capsule 3oguest. Thus, these hosts are very rigid and thus highly sensitive to small perturbations in guest size/shape/electronics. This makes these systems really useful for studying recognition processes. [twist figure]
Chapman, R. G.; Sherman, J. C. J. Am. Chem. Soc. 1995, 117, 9081-9082.
Templation of a Hemicarceplex (Darren Makeiff, Doug Pope) Hemicarceplexes retain their guests through chromatography and other routine conditions, but they can allow guests to escape without rupturing covalent bonds, given sufficient heat and time. In the formation of hemicarceplex 4oguest, capsule 3oguest (see Early Work) can form, in principle. Is this capsule a good model for the transition state of the GDS in forming 4oguest, as it is for forming 2oguest? A template study was performed and the results show that there is no correlation between the two systems. The best templates for forming 4oguest are para-disubstituted benzenes. Thus, in the transition state of the GDS for 4oguest, the cavity is much larger than that for forming 2oguest. From the data obtained and examination of CPK models, it is apparent that the GDS is formation of the third or fourth bridge. At this point the bowls are held rigidly apart, which creates a large cavity. Models also make it clear that guest exchange is still plausible at the three-bridged stage, as there remains one large portal. Another relevant point is that the yields in this reaction are terrible, never above 30%, and often below 20%, which is much lower than the yields for 2oguest. This is consistent with the formation of the second bridge proceeding in the absence of a template effect. Thus polymerization can readily ensue in this case, while for carceplex 2oguest the parts are preorganized from the start, by the template and the inter-bowl hydrogen bonds, to promote formation of the carceplex.  Makeiff, D.; Pope, D.; Sherman, J. C. J. Am. Chem. Soc. 2000, 122, 1337-1342. Template Effect with no Hydrogen Bonds (Ayub Jasat) The cavity of carceplex 7oguest is roughly the same size and shape as that of 2oguest, perhaps 10% larger. The template effect on forming carceplex 7oguest from reaction of benzylthiol-cavitand 6 with benzylbromide-cavitand 5 addresses the question: what happens in forming a carceplex with a cavity roughly the size in 2oguest, but without the possibility for a hydrogen bonded capsular precursor? Our hypothesis was: If the two bowls cannot associate, there should not be much of a template effect. Contrary to our prediction, we have found a large template effect at play. There is no correlation between the guest-dependence of the template effects for the two carceplex systems. We've made the following conclusions: (1) The GDS must be formation of the third bridge. The portals are too small to allow guest exchange after the third bridge is formed (according to CPK models). (2) The cavity of the transition state of the GDS is well defined and held rigidly, thus giving a large range in template ratios. (3) The size/shape/electronics of the cavity of 7oguest is different from that of 2oguest. The small difference is obviously significant as the guest-selectivity is vastly different. Thus again, small perturbations in a system can be manifested by large effects in the energetics. Again, this makes the cavitand system in general very useful for probing subtle noncovalent interactions.  Bryant, J. A.; Blanda, M. T.; Vincenti, M.; Cram, D. J. J. Am. Chem. Soc. 1991, 113, 2167?2172.
Large Vessels that Entrap Multiple Molecules (Naveen Chopra, Darren Makeiff) We are currently working toward investigating the use of larger guests, and more interestingly, multiple guests as templates. We have prepared a large trimer carceplex, 12o(DMF)3, whose synthesis involves bis-benzylation of tetrol 1 (available in 10 gram quantities) in opposing "A,C" positions to give diol 2 (20% yield), followed by cyclization to trimer 7 (40% yield). Removal of the benzyls followed by capping using 1,3,5-tris-bromomethylmesitylene in DMF in the presence of Cs2CO3 and KI yields 12o(DMF)3 in 40% yield. (Methyls have been omitted from the mesitylenes of the drawings of 12 for clarity.) This is a very large carceplex, considering that all previous carceplexes only entrap one molecule of DMF. In addition, no carceplex or hemicarceplex has been reported that contains three guest molecules of any kind. DMF cannot escape 12 even after prolonged heating in solution; thus 12 is indeed a carceplex and contains only very small pores. Variable temperature experiments reveal that the DMFs reside in three unique environments, and the overall environment is a cross between the gas and liquid states. In preliminary results we have found that the formation of carceplex 12 can be templated by single molecules, a pair of molecules, and sets of three molecules. Competition experiments suggest that single molecules are better templates that multiple molecules, at least at higher temperatures. It appears that multiple molecules may be favored at lower temperatures, which indicates that three molecules may be better able to complement the nooks and crannies of the cavity (better van der Waals contacts, higher enthalpy of complexation) but the entropy of organizing three molecules must be overcome (at lower T).   Chopra, N.; Sherman, J. C. Angew. Chem. Int. Ed. 1997, 26, 1727-1729.
Current Large Vessel Work (Darren Makeiff) In current work, we have prepared a carceplex made of six cavitands, which has a cavity volume of about 900 Ĺ3. This is more than double the volume of trimer carcerand 12, which is also evident by the entrapment of seven molecules of DMSO. The reaction appears to manifest high guest-selectivity in forming the six-cavitand carceplex, and high selectivity in the number of guests as well. [n]Cavitands and [n]Carceplexes (Christoph Naumann, Samuel Place, and Jianyu Sun) We have prepared a new series of cavitands that contain more than four resorcinol subunits. These compounds have new symmetry and new cavity size and shape, and thus have the potential to be used to create novel hosts. For example, pentathiol [5]cavitand 13, in DMF in the presence of base and atmospheric oxygen gives [5]carceplex 14o(DMF)2. This is the first carceplex or hemicarceplex linked via disulfide bonds, which opens the door to new chemistry such as reversible or irreversible formation, depending on conditions. Moreover, one can envision a releasing mechanism via reduction of the disulfides. Such release may find application in drug delivery.  Naumann, C.; Roman, E.; Ren, T.; Peinador, C.; Patrick, B.; Kaifer, A. E.; Sherman, J. C. Chem. Eur. J. 2001, 7, 1637.
Vessels with Multiple Chambers (Naveen Chopra, Christoph Naumann, Rajesh Mungaroo) We have created higher order structures, such as bis-carceplex 15, obtained from tetramer 11. As we also have hexamer 16 in hand, we preliminary evidence for the corresponding tris-carceplex, 17o(guests)3. Such compounds have the potential to exhibit communication between guests in adjacent but physically separated chambers.  Chopra, N.; Sherman, J. C. Angew. Chem. Int. Ed. 1997, 26, 1727-1729.
Potential Uses of Large Vessels We have photolyzed 12obutyrophenone in an attempt to create a enol stabilized as a result of being cut off from a proton source and thereby unable to tautomerize into a ketone. Photolysis experiments yield a compound whose NMR spectrum is consistent with entrapped ethylene and the enol form of acetophenone. Thus, we have been successful in stabilizing the enol. This vessel is useful for stabilization of reactive intermediates never before stabilized in a carceplex or hemicarceplex. Potential uses for these vessels as reaction chambers or for novel multiple molecule template studies are plausible. Unique investigations such as revelation of novel physical properties (e.g., effective phase) of the encapsulated species are possible. Such studies may help bridge the gap between single molecule effects and bulk solvent effects: When does a group of molecules become a bulk solvent and constitute a phase? Can several molecules organize themselves via a microcrystallization-like process and act as a singular entity? These questions are fundamental to myriad processes such as formation of monolayers and liquid crystals, and to assembly of membranes and other macromolecular assemblies such as nucleosomes. Studies on clusters of atoms and molecules strive to elucidate the structure and dynamics of small groups of molecules held together loosely via weak noncovalent interactions. Such studies are difficult to perform due to the instability of the structures and limitation to dilute samples (clusters are usually studied in the gas phase or on surfaces). We will be able to study clusters as homogeneous, stable entities at high concentrations in solution or in the solid state. Thus, our compounds will facilitate studies of clusters, which have remained somewhat elusive to thorough analysis. Clusters in a carceplex could lead to a net alignment of molecules with a net dipole. Such a large molecule with a net dipole could manifest unique properties and could lead to materials applications such as ferroelectric materials. A somewhat related question is whether the collection of molecules function initially as a template or as a kind of solvent effect, or is there no difference? Thus, studies of encapsulation of multiple molecules will not only lead to a variety of novel investigations, they should have an impact on related processes in materials science and in the biological areas related to health. Other potential applications include use as drug delivery devices, sensors, micro-reaction chambers, and removal of toxins from waste-water. For example, a drug could be incarcerated (adamantidine has already been), targeting groups could be linked to the vessels to bring them to the desired tissues, release mechanisms could be incorporated such as enzymatic hydrolysis of ester linkages or photolysis of photolabile linkers. Naturally, issues such as solubility, crossing the blood-brain barrier (polarity is highly tunable), and toxicity would require investigation. De Novo Proteins (Bruce Gibb, Adam Mezo, Ashley Causton, Diana Wallhorn, Emily Seo, Heidi Huttunen, Jon Freeman) De novo proteins are proteins designed from first principles, as opposed to natural proteins, or mutants of natural proteins. De novo proteins offer chemists and biochemists unique opportunities to understand and predict the structure of newly discovered proteins, and to perform functions that may be inaccessible with natural proteins or enzymes. The problem in designing de novo proteins is that there are far too many conformations available to polypeptides within a small energy range to predict structure with any confidence. Our approach toward de novo protein design largely circumvents the multi-minima problem by drastically reducing the number of available conformations. We do this by taking advantage of our ability to synthesize molecules, and in particular, we exploit the high rigidity and well defined structure of organic hosts such as cavitands, which can act as scaffolds. Such hosts can be used both for their hydrophobic binding pockets and as scaffolds to preorganize poly-peptidic units into a tertiary protein structure. Peptides are designed that will fold into alpha-helices that contain a hydrophilic and a hydrophobic face. Once attached to the scaffold the peptides form well-defined helical bundles. The problem with de novo design is control. How can you be sure the structure will be helical? How can you be sure four helices will be involved, not three or five? How can you control the arrangement of the helices, parallel versus anti-parallel? Use of a rigid organic scaffold and organic synthesis, we solve these problems. The only remaining challenge is to create monomeric native-like structures. This means there should be no aggregation of the proteins, and their parts should be conformationally specific; there should be one predominant conformation.  We have designed peptide sequences that, when folded into an alpha-helix, will present hydrophobic and hydrophilic faces. For example, EELLKKLEELLKKG (E = glutamic acid, K = lysine, L = leucine, G = glycine) positions the five leucines (i.e., five hydrophobic sec-butyl groups) on one face. The other face contains the salt-bridged pairings of the positive and negative charges of the glu and lys. We can link the peptides to the organic scaffold via a variety of linkages. Typically, we chloro-acetylate the N-terminus of the peptides and react this electrophilic group selectively with thiols from our cavitands or other templates/scaffolds. Alternatively, cysteines incorporated into the peptide allow us to attach the peptides at any position in the sequence, for example via disulfides formed with the template. The peptides are readily made on our automated peptide synthesizer, cleaved from the resin, purified, and then coupled to the template. The ensuing protein is then purified and characterized. Most of our initial information comes from circular dichroism (CD), which crudely, is like a UV spectrum of chiral moieties. CD gives a measure of overall helicity. CD is a secondary structural determinant, and tells nothing of the tertiary structure or packing of the protein. We measure stability by monitoring the CD spectra in the presence of denaturants such as guanidine hydrochloride. We have found the bulk of our de novo proteins to be helical and highly stable. The final question then is native-like structure. Do the proteins exist in single conformations? This is not only a challenge to design in, it is also difficult to diagnose. One method to explore native-like structure is NMR. Conformational specificity is marked by sharp signals, and a high dispersion of signals. This we have indeed seen in several of our proteins. Another method to probe for conformational specificity is exchange of N-H groups with D2O. This can be done easily by NMR simply by dissolving the protein in D2O and watching the NH signals disappear over time. We have had some NHs linger for weeks, which is on par with natural proteins. All in all, some of our best designs appear to be highly stable and native-like. Current efforts are toward further understanding what makes a protein native-like, making less stable proteins, which are more like natural proteins, making anti-parallel bundles, and making catalysts. As for catalysts, our systems have great potential because we can design in virtually any functional group wherever we want, and our proteins are highly stable at high temperatures, where reaction rates will be enhance further still. Adam R. Mezo, and John C. Sherman, J. Am. Chem. Soc., 1999, 121, 8983-8994.
|
 |
