
Current Tanner Research Projects/Group Members updated Oct. 8 '03
UDP-N-Acetylglucosamine 2-Epimerase.
Wayne Chou, B.Sc. UBC, Ph.D. Candidiate, e-mail: wkchou@interchange.ubc.ca
Andrew Murkin, B. Sc. UBC, Ph.D. Candidate, e-mail: murkin@chem.ubc.ca
The bacterial UDP-N-acetylglucosamine 2-epimerase catalyzes the reversible interconversion of UDP-N-acetylglucosamine (UDP-GlcNAc) and UDP-N-acetylmannosamine (UDP-ManNAc) (Figure 1). This provides bacteria with an activated form of ManNAc residues that can be used in the biosynthesis of a variety of cell-surface polysaccharides. In several pathogenic strains, such as Streptococcus pneumoniae types 19F and 19A, ManNAc residues are found in the antiphagocytic capsular polysaccharide that protects the bacteria from the immune system of the host.
Figure 1. Reaction Catalyzed by the Bacterial UDP-GlcNAc 2-Epimerase.
The mammalian UDP-GlcNAc 2-epimerase catalyzes the formation of UDP and ManNAc from UDP-GlcNAc (this is essentially irreversible and technically not an epimerization) (Figure 2). The ManNAc formed in this process is ultimately converted into N-acetylneuraminic acid 9-phosphate that serves as a precursor to the sialic acids in mammals. The sialic acids are located on the distal side of glycan chains in many cell surface glycoconjugates and play important roles in mediating cellular interactions. In human cells it has been demonstrated this enzyme catalyzes the rate-limiting step of sialic acid biosynthesis and thus dictates the extent of cell surface sialylation.
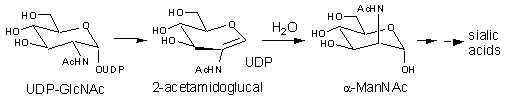
Figure 2. Reaction Catalyzed by the Mammalian UDP-GlcNAc 2-Epimerase
Studies on the UDP-GlcNAc 2-epimerase from E. coli support a mechanism involving an initial anti-elimination of UDP to generate the intermediate 2-acetamidoglucal, followed by a subsequent syn-addition of UDP to give the product UDP-ManNAc, J. Am. Chem. Soc. 1997, 10269. (Figure 1). Solvent isotope incorporation studies and kinetic isotope effect measurements show that the proton at C-2 is removed and replaced in a non-stereospecific manner. Positional isotope exchange (PIX) experiments with 18O-labeled UDP-GlcNAc show that the C1-O bond is transiently cleaved and implicate UDP as an intermediate during catalysis. It has also been shown that upon extended incubations the intermediates UDP and 2-acetamidoglucal are occasionally released from the active site and accumulate in solution. Similar studies with the mammalian enzyme support a mechanism involving the anti-elimination of UDP, followed by the syn-addition of water, J. Am. Chem. Soc. 2003, 2455. (Figure 2).
Peptide Epimerase from Spider Venom.
Andrew Murkin, B. Sc. UBC, Ph.D. Candidate, e-mail: murkin@chem.ubc.ca
Ribosomally produced peptides that contain D-amino acids have been isolated from a number of vertebrate and invertebrate sources. In each case, the D-amino acids are introduced by a posttranslational modification of a parent peptide containing only amino acids of the L-configuration. The only known enzyme to catalyze such a reaction is the peptide epimerase (also known as peptide isomerase) from the venom of the funnel web spider, Agelenopsis aperta. This enzyme interconverts two 48-amino acid long peptide toxins, 1 and 2, that differ only by the stereochemistry at a single serine residue (Figure 3).
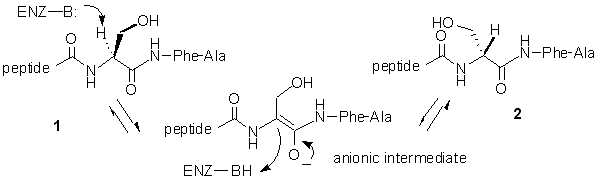
Figure 3. Reaction Catalyzed by the Peptide Epimerase.
In a recent paper we reported the synthesis and testing of two pentapeptide analogs that contain modified amino acids at the site normally occupied by the substrate serine residue, J. Org. Chem. 2002, 8389. When the L-chloroalanine-containing peptide 3 was incubated with the epimerase it was converted into the dehydroalanine-containing peptide 4 via an elimination of HCl (Figure 4). The dehydroalanine peptide 4 was independently synthesized and found to act as a potent inhibitor of the epimerase (IC50 = 0.5 mM). These results support a direct deprotonation/reprotonation mechanism in which a carbanionic intermediate is formed (Figure 3). The observed inhibition by 4 can be attributed to the sp2-hybridization of the a-carbon in the dehydroalanine unit that mimics the planar geometry of the anionic intermediate.
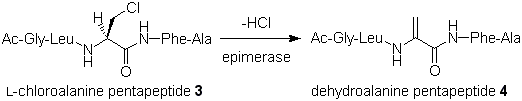
Figure 4. Reaction of Chloroalanine Pentapetide with Peptide Epimerase.
ADP-Heptose 6-Epimerase.
Jay Read, B.Sc. Okanagan University College, Ph.D. Candidate, e-mail: jread@chem.ubc.ca
James Morrison, M.Sc. University of Victoria, Ph.D. Candidate, e-mail: jmorrison@chem.ubc.ca
Dr. Raef Ahmed, Ph.D. Cambridge University, postdoctoral researcher, e-mail: rahmed@chem.ubc.ca
The enzyme ADP-L-glycero-D-manno-heptose 6-epimerase (ADP-heptose 6-epimerase) catalyzes the interconversion of ADP-D-glycero-D-manno-heptose and ADP-L-glycero-D-manno-heptose (Figure 5). The latter sugar nucleotide is a required component for the construction of the inner core of the lipopolysaccharide in most Gram-negative bacteria. Bacteria lacking this activity exhibit a truncated lipopolysaccharide structure, a decreased pathogenicity, and an increased permeability to hydrophobic antibiotics. Thus inhibitors targeting this enzyme could be co-administered with a wide variety of antibiotics and serve to greatly enhance the efficacy of the drugs.
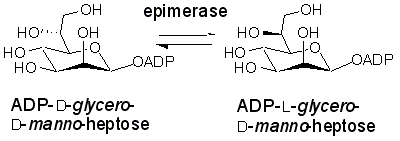
Figure 5. Reaction Catalyzed by ADP-Heptose Epimerase
The mechanism employed by this epimerase is of interest to enzymologists since the enzyme acts at an unactivated center (one lacking a relatively acidic C-H bond). It has been demonstrated that the enzyme uses a transient oxidation/reduction strategy employing a tightly bound NADP+, however little other mechanistic information is available. There are at least four quite distinct, yet quite reasonable mechanisms that could be postulated for this reaction: A) oxidation/reduction directly at C-6, B), oxidation at C-4 (dehydration rehydration), C) oxidation at C-4 (retroaldol aldol), D) oxidation at C-7 (deprotonation/reprotonation) (Figure 6). Experiments using deoxy substrates, fluorinated substrates, and isotopic labels are currently being used to differentiate between these possibilities.
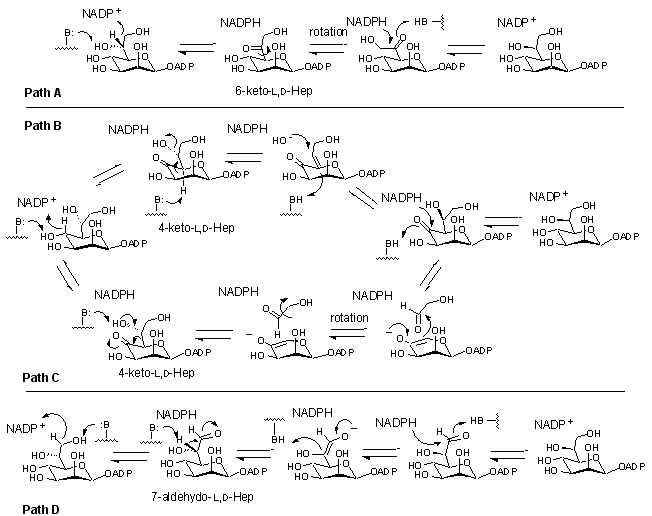
Figure 6. Potential Mechanisms Employed by ADP-Heptose Epimerase.
Sialic Acid Synthase.
Dave Simard, B.Sc. Dalhousie, Ph.D. Candidate, e-mail: dsimard@chem.ubc.ca
Sialic acid synthase catalyzes the third step in the biosynthesis of the sialic acids and condenses phosphoenolpyruvate (PEP) with ManNAc 6-phosphate (ManNAc6P) to give neuraminic acid 9-phosphate (Neu5Ac9P) (Figure 7). Sialic acids play key roles in mediating cellular interactions in humans, and controlling their biosynthesis could have profound implications in a variety of disease states. Two general mechanistic strategies can be invoked for this reaction. The first involves the generation of an enolate by the dephosphorylation of PEP, followed by an attack of the enolate on the open chain form of the sugar (Figure 7, Path 1). A second mechanism involves the attack of the C-3 carbon of PEP onto the aldehyde of ManNAc6P (Figure 7, Path 2). The addition of water at C-2 followed by the loss of phosphate and subsequent ring closure could then generate the product. The first two steps of this reaction would likely proceed in a stepwise manner, involving an oxocarbenium ion intermediate. Isotopic labeling studies are currently underway that will discern between these mechanisms and will uncover the stereospecificity of the PEP addition.
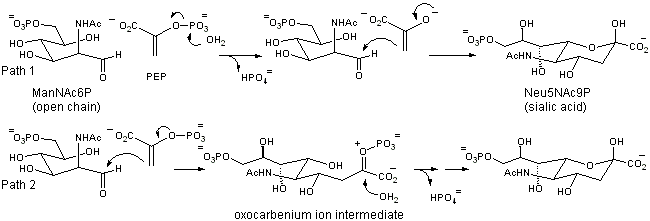
Figure 7. Two Potential Mechanisms for the Reaction Catalyzed by Sialic Acid Synthase